Une nouvelle stratégie de simulation pour prédire la dynamique de molécules complexes
Simuler la dynamique quantique des molécules complexes est un enjeu pour la compréhension de tous les systèmes organiques, enjeu qui devient rapidement un défi numérique à croissance exponentielle avec le nombre d'atomes dans la molécule. Les chercheurs ont mis au point une nouvelle stratégie, utilisant des méthodes d'apprentissage automatique et d'analyse de l'intrication pour réduire par plus d'un facteur un million les capacités de calcul nécessaires.
L'excitation d'une molécule par un photon peut conduire à la libération d'électrons comme pour l'effet photovoltaïque ou à des réactions telles qu'une rupture de liaison ou un changement de structure. La dynamique réactionnelle passe non seulement par le mouvement des électrons qui sont directement excités et qui constituent le système proprement dit, mais aussi par le mouvement de tous les atomes environnants, appelé bain, à travers des couplages entre les mouvements électroniques et atomiques. Les premières étapes qui orientent la réaction ont lieu à des temps très courts, picosecondes ou femtosecondes. Elles mettent en jeu une compétition entre d'une part, des couplages particuliers conduisant à des mouvements cohérents qui contribuent à la réaction, et d'autre part, des couplages indifférenciés conduisant à une répartition statistique de l'excitation dans le bain et qui contribuent à la dissipation de l'énergie. Décrire de façon exhaustive tous les couplages est le plus souvent prohibitif en temps de calcul mais d'un autre côté, traiter les couplages uniquement de façon statistique ne permet pas de rendre compte correctement de la réaction. Dans ce travail, grâce à une collaboration menée par un chercheur de l'Institut des nanosciences de Paris (CNRS/Sorbonne Université) et des chercheurs britanniques, une nouvelle stratégie a été élaborée pour traiter ce problème complexe, en l'appliquant à la simulation de la dynamique d'une réaction particulièrement prometteuse pour améliorer le rendement de cellules photovoltaïques. Lors de cette réaction, l'excitation par la lumière d'un état électronique d'une molécule dimère (figure a) est transférée à deux autres états électroniques, provoquant ainsi un dédoublement de l'excitation. Ces nouvelles simulations ont été confrontées avec succès à des expériences de spectroscopie ultra-rapides, démontrant qu'elles rendent compte correctement du détail des mécanismes réactionnels. Elles sont généralisables à d'autres réactions photo-induites et pourraient donc être exploitées pour la conception de nouveaux systèmes moléculaires plus efficaces.
Le point de départ de la simulation est la description complète, par des calculs de type ab initio couramment utilisés pour la description quantique des molécules, du système électronique et des vibrations atomiques, au nombre de 252 pour la molécule étudiée, en prenant en compte tous les couplages. Les chercheurs ont ensuite appliqué une méthode d'apprentissage automatique (K-means clustering) pour grouper les modes de vibration en fonction de la similarité de leurs propriétés de symétrie et de couplage (figure b). Le système électronique est alors couplé avec un nombre limité de groupes (7). Une fois cette structure sous-jacente révélée, l'ensemble des états intervenant dans la réaction peut être efficacement représenté à l'aide d'une structure mathématique appelée réseau tenseur (tree tensor network) qui possède ici 7 branches de tenseurs, chaque branche regroupant les modes de vibrations similaires. La croissance de la dimension du problème à traiter avec le nombre d'atomes devient polynomiale au lieu d'exponentielle. Ce réseau tenseur prend en compte tous les liens dus aux différents couplages et reste encore extrêmement consommateur de mémoire. Les chercheurs ont finalement appliqué une méthode d'analyse de l'intrication qui, par l'optimisation de l'entropie des différents liens entre tenseurs, recombine le réseau de telle sorte que les couplages soient regroupés et que les liens soient simplifiés au minimum (entanglement renormalization). Ceci constitue l'étape clé pour réduire énormément les temps de calcul et rendre ainsi la simulation d'un système complexe réalisable, en extrayant à partir d'une description complète l'information essentielle sur les cohérences entre les mouvements.
Cette stratégie peut être envisagée beaucoup plus généralement pour tout système quantique ouvert sur un bain et a donc potentiellement un champ d'application très vaste.
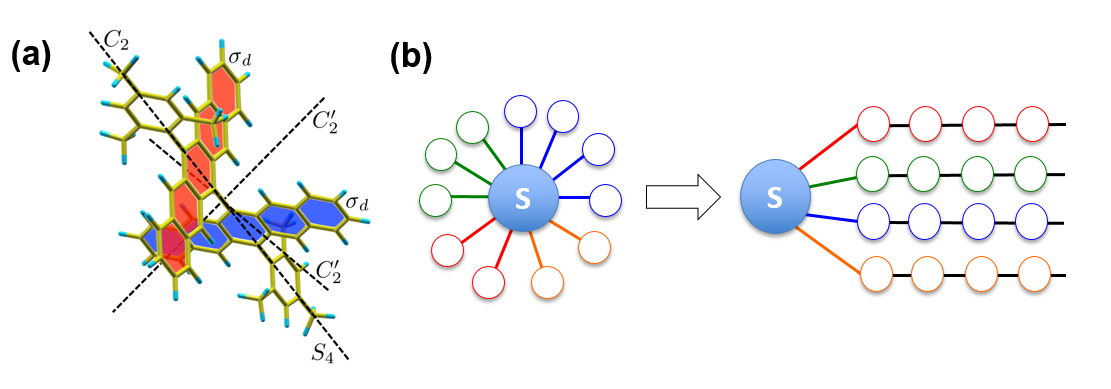
(b) Représentation schématique du système électronique (disque bleu S) couplé via les traits colorés avec les différents modes de vibrations et du regroupement effectué en plusieurs branches par la méthode K-means en fonction de la symétrie des modes et de la force des couplages. S devient couplé à un petit nombre de branches qui forme la structure du réseau tenseur.
Références
Tensor network simulation of multi-environmental open quantum dynamics via machine learning and entanglement renormalisation. Florian A.Y.N. Schröder, David H.P. Turban, Andrew J. Musser, Nicholas D.M. Hine et Alex W. Chin, Nature Communications, le 5 mars 2019. DOI: 10.1038/s41467-019-09039-7
Consulter l’article sur la base d’archives ouvertes HAL.
A molecular movie of ultrafast singlet fission. Christoph Schnedermann, Antonios M. Alvertis, Torsten Wende, Steven Lukman, Jiaqi Feng, Florian A.Y.N. Schröder, David H.P. Turban, Jishan Wu, Nicholas D.M. Hine, Neil C. Greenham, Alex W. Chin, Akshay Rao, Philipp Kukura et Andrew J. Musser, Nature Communications, le 16 septembre 2019. DOI: 10.1038/s41467-019-12220-7